1. 細胞標識
細胞性粘菌の細胞標識には,赤い色素prodigiosinを合成する細菌Serratia marcescensを餌とする方法 (Raper, 1940),neutral redなどの塩基性色素による生体染色 (Bonner, 1952),遺伝マーカーを用いる方法 (Bonner, 1959; Leach et al., 1973),放射ラベルされたチミジンなどによる標識 (竹内・佐藤, 1965; Tasaka and Takeuchi, 1978; Pogge von Strandmann and Kay, 1990),蛍光色素による生体染色 (Springer and Barondes, 1978),lacZレポーター (Buhl and MacWilliams, 1991),green fluorescent protein (GFP) (Zimmermann and Siegert, 1998),などが用いられてきた.Serratia marcescensで増えた細胞では,neutral redと同様に予定柄細胞がより強く染色されるが,この方法は十分な色素量を維持するのに技術を要することから,より簡単な色素による直接染色にすぐにとってかわられた.
遺伝マーカーや放射標識は確実な方法だが,結果を得るのに手間がかかるので,最近ではあまり用いられない.lacZレポーターは,actin15のようなすべての細胞で発現する遺伝子のプロモータを用いると、組織中の個々の細胞の由来を同定でき、発現が一部の細胞に限られる遺伝子のプロモータを用いると、標識された細胞のうちその遺伝子を発現している細胞の同定ができる.しかし、細胞の固定を要するので経時的な観察ができない.これに対して,生体染色やGFPを用いると,生きている状態での変化を見ることができるのが最大の長所だといえる.GFPについては前節で扱われているので,ここでは色素による生体染色を取り上げ,その原理と実際の方法を述べる.
Neutral red, Nile blue, methylene blueによる生体染色
Neutral redまたはNile blue, methylene blue (図1)で染色された細胞は,白色光のもとでそれぞれ赤あるいは青に見えるが、これは細胞内の酸性の小胞に塩基性色素が蓄積することによる.いくつかの染色方法が記載されているが (Sternfeld and David, 1981; Yamamoto and Takeuchi, 1983; Bonner et al., 1990; Sternfeld, 1992),次のようにまとめることができる.
細胞の懸濁液に終濃度が0.002〜0.005\% (methylene blueの場合はこの5倍濃度)程度になるように色素のストックを加えて撹拌し,室温で短時間(数秒)または氷冷中で数分置いたあと遠心操作で色素液を除き,2回ほど細胞を洗う.染色していない細胞と混合する時は、混合の前に2時間ほどバッファー中で培養し、その間に細胞から吐き出された色素を除くため再び洗浄する.溶液のpHが高いほど有効な色素濃度は高くなり,またメーカーによる純度の違いもあるので,使用する濃度は自分で決める必要がある.水溶性色素なので,ストックは蒸留水に終濃度の10〜20倍の濃度になるように固体の色素を溶かし,0.2〜0.45 μmの無菌フィルターを通してから滅菌した容器に保存する.
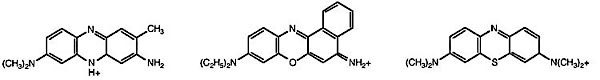
図1 Neutral red, Nile blue, methylene blueの構造.
増殖期の細胞を集めて染色する場合が多いが,多細胞期の細胞を分散して染色すると,よりはっきりした染めわけができることがある.また,増殖用の培地に上述の濃度の1/10程度の色素を入れておくことでも染色できる.増殖期の細胞では,これらの色素は食胞に蓄積し,染色の強さは細胞ごとに異なる.染色の強さはその後減少し,強く染まった細胞の数も減るが,どの時期でも予定柄細胞と同じ程度の染色が見られるので,予定柄細胞が染まるというより,予定胞子細胞だけで染色が薄くなる.予定柄細胞では、自食胞にこれらの色素が蓄積し,その自食胞が融合して大きくなるので,染色された顆粒の大きさを注意深く見ることで予定柄細胞とそれ以前の細胞をある程度区別することができる.自食胞が十分発達してくると,染色をしなくても位相差顕微鏡で予定柄細胞を容易に識別できる.
予定柄領域で濃く,予定胞子領域では薄いという染色パターン (Bonner, 1952; MacWilliams and Bonner, 1979; Sternfeld and David, 1981)が現れる時期は,実験条件によって大きく変わり,早い場合は突起の現れる前に集合体の中央部に一時的に強く染まる細胞が集まることがあり (Carrin et al., 1996),遅ければ移動体がしばらく移動してからはじめて明瞭になる(Bonner, 1952; Bonner et al., 1990).山本によると,塩濃度の高い環境 (40 mM Na2HPO4 / KH2PO4, pH 6.4, 1.5 g/l KCl)で発生させると染色パターンは突起形成の頃からあらわれる (Yamamoto and Takeuchi, 1983).分化のパターンが生じているのに移動体全体が一様に染まって見えることがあるのは,Bonner らが示唆しているように予定胞子細胞の状態の違いを反映していると考えられる.特にアンモニアの影響が大きい.
アンモニアのような弱塩基の作用を考察するには,脂質膜で隔てられたコンパートメント間の弱塩基の移動について考えておく必要がある.上述の染色の条件では,これらの色素はおそらく電気的に中性の形で膜を透過し,細胞質でその一部がプロトンと結合して正電荷を得ると考えられる.細胞質にある中性の分子は,自食胞のような酸性の小胞に入ると,多量にあるプロトンと結合してほとんどすべてが正電荷を持ち,膜を透過しにくくなるのでそこに蓄積する.濃縮の度合いは,膜の両側のプロトン濃度の比にほとんど比例する.
さらにneutral redは中性付近のpH指示薬でもあり,アルカリ性では黄色だが酸性で濃い赤色となるため ( pKa = 6.8 ),自食胞は濃い赤色に染まり,その結果,予定柄細胞は予定胞子細胞に比べて赤く見えることになる.しかし,細胞質にも濃度は低くてもかなりの量の色素が残るので,細胞質pHが低下する条件では予定柄細胞も予定胞子細胞も細胞質全体が赤色がかって見える.
一方,アンモニアも弱塩基なので酸性小胞に集まり、その量が多いと酸性小胞のpHを高くして細胞質pHとの差を縮める.このような条件では塩基性色素は酸性小胞には蓄積せず,また小胞内部のpHも高いので,強い染色はおこらない.既に自食胞に多くの色素分子が蓄積しているときにアンモニアの濃度が上昇すると,細胞質の色素濃度が急激に上昇する.したがって,アンモニアの濃度の上昇によって自食胞の色は薄くなるが細胞質の色が濃くなるということがおこりうる.
このように,neutral redなどによる細胞内の染色パターンの生成はかなり複雑な現象なので,移動体における染色パターンにもpHや塩濃度,局所的な細胞密度,プレート全体の細胞数,など様々な要因によって影響を受ける.
蛍光色素による生体染色
一方,fluorescein isothiocyanate (FITC) のような蛋白質に結合する蛍光色素や,カルボシアニン色素のような脂溶性の蛍光色素は,細胞の分化に関わり無く細胞の膜系を染色するので,キメラを作るとき細胞の由来を示す標識として用いられたが (Springer and Barondes, 1978; MacDonald and Durston, 1984; Akiyama and Inouye, 1987),最近は細胞質全体を標識できる他の色素やGFPに取って代わられ,以前ほど用いられない.Fluoresceinのchloromethyl誘導体に膜透過性を賦与したchloromethylfluorescein diacetate (CMF-DA, Molecular Probes, C-2925)は,細胞質に入ってから蛋白質に結合するので,かなり安定で均一な細胞質標識を得ることができる (Knecht and Shelden, 1995; Tsujioka et al., 1999).また,デキストランに結合させた蛍光色素は,エレクトロポレーションなどで細胞内に入れることによって,非常に安定した標識となる.一般的なエレクトロポレーションの装置を使う限り少量の細胞しか扱えないが、個々の組織中での細胞の分布を見るような実験には、10^6個ほどの細胞で足りることが多い.この他に、様々な細胞内構造を選択的に染色する蛍光色素が知られており、標識を兼ねて使える場合がある.
CMF-DAによる染色
固体のCMF-DAを、乾燥したdimethyl sulphoxide (DMSO)に5 mg/mlの濃度で溶かしてストックとする.1回の実験に少量の細胞しか使わない場合は,50 μg/ml に小分けして売っているチューブ (Molecular Probes, C-7025)にDMSOを 10 μl加えて溶かすのが無駄が少ない.ストックは-80℃で保存するのが安全だが,容器に乾燥した窒素を吹き込んでからすぐに蓋をし,そのまま-20℃ で保存してもよい.
染色には10^7 cells/ml以下の濃度の細胞懸濁液に1/100量のストックを加え,室温暗所で10〜30分振盪する.エッペンドルフチューブの中で少量の細胞を染色する場合は,チューブを横倒しにしておき,ときどき撹拌するのでもよい.2〜3分たつと酢酸エステルが加水分解されて蛍光を発するようになり、細胞懸濁液が黄色くなるのがわかる.遠心で色素溶液を除き,2回ほど細胞を洗浄する.他の細胞と混合するときは,洗浄した細胞をさらに少なくとも1時間ほど暗所で振盪し,再び細胞を洗う.
デキストラン結合色素による染色
色素はエレクトロポレーションで細胞に導入する \footnote{ Scrape-loadingによるデキストラン結合色素の導入も行なわれてい る (Schlatterer et al., 1992).}
様々なエレクトロポレーションの方法が公表されているので (Howard et al., 1988; Abe et al., 1988; van Haastert et al., 1989; Schoen et al., 1989; Yumura et al., 1995; Knecht and Pang, 1995),それらを元に目的にあった方法をみつければよいが,その際に考慮すべき点がいくつかある (葛西・稲葉、1986; Inouye, 1998b).同じ条件でも細胞膜に孔のあく効率や孔の大きさは細胞によって異なる.したがって、少数の細胞にだけ色素が導入されればよい時は比較的緩やかな条件(低い電圧など)が使えるが、すべての細胞を標識したい場合は半数ほどの細胞が壊れるほどの厳しい条件が必要になることもある.特に、小さな細胞ほど穿孔の効率が低いので、細胞のサイズにばらつきが大きい時は小さな細胞が選択される可能性がある.つまり、緩やかな条件では穿孔効率の高い細胞、厳しい条件では穿孔効率の低い細胞を選択的に見る可能性があるので、そのような場合には適切なコントロールをとることが重要になる.
一方、温度も重要な要素になる.低温では穿孔の効率、孔の修復速度の両者とも低くなる.孔の修復に時間がかかるほど色素が拡散で細胞に入る時間も長くなるが、細胞にとって修復は速い方が良いと思われるので、室温で色素の導入が十分であれば冷やさない方が良い.また、イオン強度を特に高くしない限りジュール熱は大きくないので、この理由からも冷却する必要はない.しかし、axenic株でpinocytosisによる色素の取り込みを完全に抑える必要があれば、低温を用いなければならない.どのような方法をとるとしても,バッファーの組成をできるだけ細胞内のイオン組成・pH・浸透圧に近くする.ここでは以下の組成のE-buffer を用いる (20 mM Na/K2 phosphate pH 7.4, 50 mM sorbitol (or sucrose), 2 mM MgSO4, 0.2 mM CaCl2).染色液は、分子量3,000 〜 70,000のデキストランに結合させた FITC, TRITC, Texas redなどを2 〜 10 mg/mlでE-bufferに溶かし,1回毎の使用量に小分けして-20℃で保存する.
電極の幅1 cm,電極間隔2 mmのエレクトロポレーション用キュベットを用いる場合,10 μl の染色液ストックに90 μlの細胞懸濁液を加えて撹拌し,直ちにキュベットに入れて(深さ5 mm),室温で電圧0.7 〜 0.8 kV (3.5 〜 4.0 kV/cm),コンデンサ容量4 μFで1回から4回 (10 〜 15秒間隔)パルスをかける.30秒から1分後に細胞懸濁液をエッペンドルフチューブに移し,遠心で細胞を色素溶液から分け,同じバッファーで2回洗う\footnote{ 高価なdextran 結合色素を使うときは,エレクトロポレーションのあと細胞を遠心で分けて溶液を回収し,遠心で壊れた細胞などを除いて凍結保存すると再使用できる.再使用の時は,体積を合わせるために新しい溶液を少し加える.キュベットも良く洗ってすぐに乾かせば再利用できる.}.
D. discoideumを使った場合、この条件で、ほとんどすべての細胞を標識することができる(図2).しかし、細胞の状態や微妙な実験条件の違いなどがエレクトロポレーション効率に影響するので、最適な条件を見つけなければならない.例えば、Polysphondyliumのように細胞が小さい時は、より厳しい条件が必要になる.以下の関係は条件を探す時の目安となる.穿孔の効率は,電圧,コンデンサ容量に大体比例し,液のコンダクタンスにほぼ逆比例する.例えば,液量を200 μl (深さ1 cm)にすると,コンダクタンスが増えるので,もう少し強い条件がいる.また、バッファーの組成もコンダクタンスに直接反映するのでエレクトロポレーションの条件に大きく影響する.つまりイオンが増えるほど強い条件が必要になる.
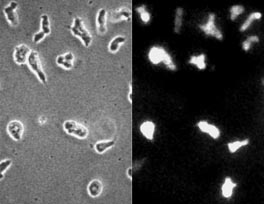
図2 エレクトロポレーションでFITC-dextranを導入した細胞.以下の条件で10秒間隔の4つのパルスを与えた(電極の幅 1 cm、電極間隔2 mm、電極電圧0.7 kV (3.5 kV/cm)、コンデンサ容量 4 μF、液量 100 μl、細胞密度1 x 10^7 cells/ml、溶液 2 mg/ml FITC-dextran (分子量70,000) in E-buffer、温度 22℃.この条件は、それぞれのパラメータについて4段階に変化させて、最も均一で明るい染色が得られたもの).約30秒後に E-bufferで15倍に希釈し、10,000 gで1秒間遠心して上清を除いたペレットを再びE-bufferに懸濁してカバーグラスにおき、約2分後に位相差と蛍光 (励起フィルタは470--490 nm、吸収フィルタは520--560 nm)の画像を記録した.画像の取得はSIT camera (C2400-08, Hamamatsu photonics)をつけたニコン倒立顕微鏡 (TMD)を用い、キャプチャボード (AG-5, Scion Corporation)をつけたパーソナルコンピュータ (PowerMacintosh 7600)にNIH Imageで取り込んだ.壊れた細胞は遠心の過程でほとんど除かれ、生き残った細胞は程度の差はあってもほとんどが色素を取り込んでいる.約15秒間隔で撮った2つの画像での細胞の形の変化から、多くの細胞はすでに活発に運動していることがわかる.細胞はAx-2、増殖培地から集めて約2時間.
細胞標識以外の応用として、デキストランに結合したpHやカルシウム感受性色素を導入するのにもこの方法が使える.色素の導入の後すぐに細胞を使いたい場合は、細胞の回復のために (例えば膜電位の回復に30分以上かかる.van Duijn et al., 1990)プラスチックシャーレの中で1時間ほど静置し,なお底に接着していない細胞がいれば、それらを洗い流してから用いるのが良い.蛍光測定や細胞外液の潅流の方法については立ち入らない (Inouye, 1998a, b).
TRITC, FITC, XRITCによる染色
Tetramethylrhodamine isothiocyanate (TRITC), fluorescein isothiocyanate (FITC), X-rhodamine isothiocyanate (XRITC)は,25〜50 mg/mlの濃度でよく乾燥したdimethyl sulphoxide (DMSO)に溶かしてストックとする.染色にはストックを100倍になるよう使用するバッファーに溶解し,溶けきれない固体があればそれを遠心で取り除いてから細胞を懸濁して,暗所で10〜30分振盪したのち細胞を洗う.
DiO, DiI, DiD, DiRによる染色
これらの色素は脂溶性が高く,はじめ細胞膜が強く染色されるが,次第に細胞内に取り込まれ,顆粒状の染色になる.いずれもストックはdimethylformamideまたはDMSOで作る (2.5〜10 mg/ml).不溶性の粒子は超音波処理で細かくできるが,このような固体と直接接触することで細胞が強く染色されるので,少数の細胞が濃く染まるという結果になりやすい.しかし、他では得られないような長い波長の励起光が使える.
染色の観察
塩基性色素で染色された細胞を白黒カメラで記録する時は、それぞれの色素の酸性での最大吸収波長 (neutral red, 540 nm; Nile blue, 640 nm; methylene blue, 665 nm) の近辺だけを通す干渉フィルタを用いると効率が良い.蛍光色素については、ここで取り上げた色素の励起波長と蛍光波長のピーク値を表1に示す.長い波長の励起光を用いる場合は、標準外の干渉フィルタとダイクロイックミラーが必要になる(例えばhttp://www.chroma.com/, http://www.omegafilters.com/を参照).
細胞に対する励起光の影響および色素の退色を少なくするため、励起光の強度と照射時間を必要最少限にとどめなければならない.Rhodamineの励起に使う540 nm付近の波長は走光性を誘起しやすく、細胞に対する影響も強い.これに対して、600 nm以上と490 nm付近は影響が比較的少ない (Poff and Hader, 1984).蛍光顕微鏡を使う場合、水銀ランプやキセノンランプからの励起光はNDフィルターで落し、シャッターを用いて必要な時にだけ照射する.450 nm程度より長い波長しか使わない場合はハロゲンランプでも光量は十分なので、その方が扱いやすく安全のためにも良い.共焦点顕微鏡の場合もできるかぎりフィルタで光量を減らし、画像の解像度・平均化も必要最低限にする.
表1 この節で取り上げた蛍光色素の最大励起波長と最大蛍光波長.比較のためにGFPも示した.GFPには異なった波長特性を持った様々な変異体が開発されている.これらの波長は色素の環境によって変化するので、大体の目安である.
| 色素 | 最大励起波長 (nm) | 最大蛍光波長 (nm) |
|---|---|---|
| Fluorescein (CMF-DA, FITC, FITC-dextran) | 494 | 519 |
| Tetramethylrhodamine (TRITC, TRITC-dextran) | 543 | 571 |
| XRITC | 572 | 596 |
| Texas Red (Texas Red-dextran) | 587 | 602 |
| DiO | 484 | 501 |
| DiI | 549 | 565 |
| DiD | 644 | 665 |
| DiR | 748 | 780 |
| GFP (wild type) | 395 | 501 |
| GFP (S65T) | 489 | 511 |
データの解析
画像の取得に撮像管を用いる場合、視野の周辺部でひずみが大きいので計測を行なう時は注意を要する.画像の処理や計測にはフリーウエアのNIH Image (http://rsb.info.nih.gov/nih-image/)が広く用いられている.充実した機能を持ち、マクロによって様々な操作を自動化できる.測定する量が少なければ、モニタにOHPフィルムなどを貼ってデータを取るという原始的な方法でも十分実用になるが、この場合も画面周辺部のひずみに気をつけないといけない.
第2章第2節 井上 敬 「実験形態学的手法と細胞の標識」 (一部改訂)
- 出版社および編者の承諾を得て掲載 -
細胞性粘菌グループ Homepage